club_bioinfo_RNAdirect
Emmanuel Labaronne
03/02/2020
Before starting an experiment
IT requirements
softwares
MinKNOW
MinKNOW carries out several core tasks:
MinKNOW utilizes an intuitive graphical user interface (GUI) and receives updates on a regular basis. This is the core software provided by Oxford Nanopore, without which the sequencing devices cannot be run. Data from MinKNOW is packaged into individual read .fast5 files (over 1 million of which can be generated by a single flow cell), which are a customised file format based upon the .hdf5 file type. These .fast5 files are then used by other downstream software.
Guppy
Guppy is a production basecaller provided by Oxford Nanopore, and uses a command-line interface. It utilizes the latest in Recurrent Neural Network algorithms in order to interpret the signal data from the nanopore, and basecall the DNA or RNA passing through the pore. It is optimiszed for running with basecall accelerators e.g. GPUs. Guppy implements stable features into Oxford Nanopore Technologies’ software products, and is fully supported. It receives .fast5 files as an input, and is capable of producing: - .fast5 files appended with basecalled information - .fast5 files that have been processed, but basecall information present in a separate FASTQ file
EPI2ME
EPI2ME™ is an onwards data-analysis platform created by Oxford Nanopore’s subsidiary company, Metrichor. It provides users with real-time analysis such as species identification, alignment workflows and other bioinformatics solutions. It is currently provided as a cloud-based analysis platform, which is initiated through the local EPI2ME Agent
Data acquisition and basecalling
Basecalling algorithms
The “Flip-flop” basecalling algorithm
The Flip-flop basecalling algorithm uses the raw signal to write out single-base transition probabilities, along with confidence levels for each called base, and the alternative possibilities. Flip-flop basecalling provides higher accuracy basecalls and better homopolymer resolution than previous algorithms from Oxford Nanopore Technologies. 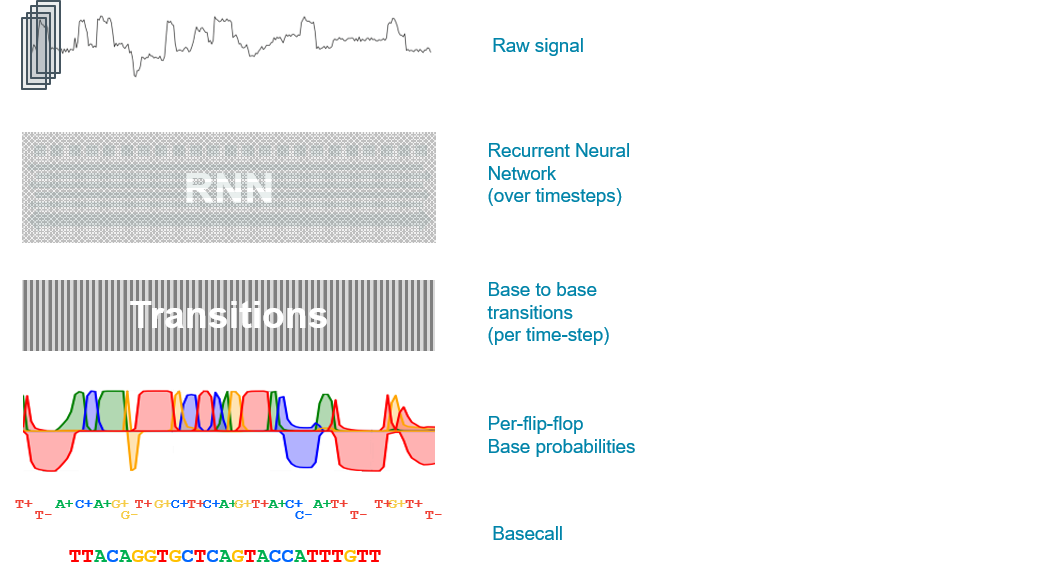
The neural network model in the Flip-flop basecaller performs label-free basecalling: instead of labelling raw data with a short sequence of bases like previous “transducer” basecalling algorithms, the model produces likelihoods of transitions between consecutive bases. The network considers two states for each base: “flip”, which is a transition from one base to the next, and “flop”, which is staying on the same base for two time-steps. The model uses Viterbi decoding to assign the likelihood of the “flip” and “flop” state for each base transition. This way, the network can distinguish between long runs of the same base (homopolymers) and multiple “stays” on the same base.
Fast vs High Accuracy models
The Guppy basecaller, which is also integrated in MinKNOW, offers two different Flip-flop models: a High accuracy (HAC) model and a Fast model. The HAC model provides a higher consensus/raw read accuracy than the Fast model. It contains a more computationally-intense Flip-flop architecture that can deliver higher accuracy using the same data produced by nanopore sequencing. It is currently 5-8 times slower than the Fast model.
The Fast Flip-flop model includes a simplified version of the Flip-flop algorithm and delivers the same level of accuracy and basecalling speed to those obtained with the older transducer basecalling algorithm, shown for comparison in the graphs below. Both models have been trained on the same datasets.
A comparison of the speed and accuracy of the two models is provided in the table and graphs below. Please note that these numbers represent the theoretical best speeds achievable with the basecallers, and a real biological sample may be basecalled more slowly.
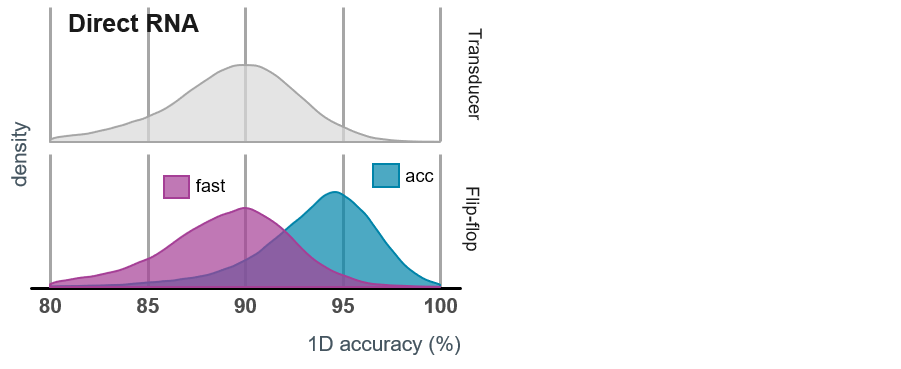
MinKNOW live basecalling: keep-up vs catch-up
Basecalling with the Fast Flip-flop model can keep up with the speed of data acquisition on all nanopore platforms. With the HAC model, basecalling on a minimum-spec laptop may not keep up with the amount of data generated. Basecalling will therefore continue after the sequencing experiment has run to completion; any reads that have not been basecalled during the experiment will be queued and processed afterwards. This is known as “Catch-up mode”.
The user therefore has two options: either to allow MinKNOW to continue in catch-up mode, or to stop the analysis and basecall the remaining reads at a later time, e.g. using stand-alone Guppy.
Stand-alone basecalling
Minimum options :
guppy_basecaller
--input_path Full or relative path to the directory where the raw read files are located
--save_path Full or relative path to the directory where the basecalled results will be saved
--flowcell flowcell_version --kit sequencing_kit version
--config configuration file containing guppy parameters
Exemple :
guppy_basecaller -i my_path/ -s RNAdirect_basecalling
--flowcell FLO-MIN106
--kit SQK-RNA002
--cpu_threads_per_caller 16
--fast5_out
To call out a list of available flow cells, kits, and config files, use the script with the –list_workflows command, outlined below:
guppy_basecaller --print_workflows
Downstream analysis
bascalling report
Based on a Rmarkdown script. You can find the script and the tutorial on the github repository : https://github.com/nanoporetech/ont_tutorial_basicqc
Mapping
For now, the best mapper for long read sequencing is minimap2. You can find the git repository here
1. Genome indexing (optional) :
minimap2 -d ref.mmi ref.fa
2. Alignement :
minimap2 -ax splice -uf -k14 ref.fa direct-rna.fq > aln.sam
or
minimap2 -ax splice -uf -k14 ref.fa direct-rna.fq | samtools -b -o aln.bam
options :
transcriptome assembly
You can build the transcriptome using STRIGTIE (version 2 or later)
stringtie -L \
-o long_reads.out_all_1.gtf \
-f 0.01 \
-j 1 \
long_reads.bam
or
stringtie -L \
-G human-chr19_P.gff \
-o long_reads_guided.out.gtf \
long_reads.bam
options :
Alternatively, you can also use FLAIR.
polyA length
There is 2 software that estimate the polyA length of the read :
1. Clone the repository :
git clone https://github.com/nanoporetech/pipeline-polya-ng
2. Usage : Edit config.yml to set the input datasets and parameters then issue:
snakemake --use-conda -j <num_cores> all
3. Input files : The input files and parameters are specified in config.yml:
- transcriptome - the input transcriptome.
- fast5_dir - directory with pass FAST5 files.
- fastq_dir - directory with the fastq files.
- summary_dir - directory with the sequencing summary files.
- spikein_fasta - (optional) fasta file with spike-inf on known poly(A) tails length. The sequence names must end in _ (for example "_50").
- min_mapping_qual - filter out reads with mapping quality less than this parameter.
- per_transcript_plots - plot the distribution of estimated tails lengths for all transcript (true or false).
- threads - number of threads to use for the analyses.
4. Output files :
- alignment/:
- aligned_reads_sorted.bam : sorted indexed alignment of reads to the transcriptome.
- input/:
- reads.fastq&ast : concatenated input reads and nanopolish index files.
- reference.fas : reference fasta (including spike-ins).
- summaries.fofn : list of sequencing summary files.
- reports/:
- filtering_report.pdf and filtering_report.tsv : nanopolish QC statistics.
- spikein_medians.tsv : expected and estimated medians of spike-ins.
- spikein_report.pdf : plots of distribution of tail lengths in spike-ins.
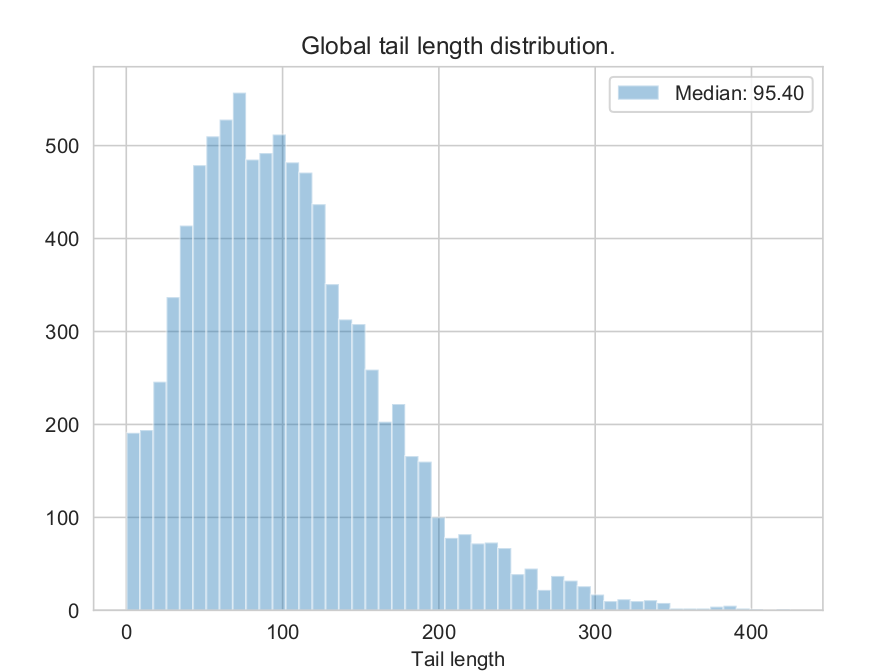
- tails_report.pdf : global and per-transcript poly(A) tail length distributions.
- tails/:
- all_tails.tsv : raw nanopolish output.
- filtered_tails.tsv : nanopolish output - PASS reads only.
- spikein_tails.tsv : results for reads mapping to spike-ins.
Exemple of tails_report.pdf :