Results
We didn’t get the PhD funding.
Object : Thèse bio-info en co-tutelle (Modolo/Bernard) visant à déterminer par des analyses multi-omiques l’impact des interactions fonctionnelles entre condensine et les nucléosomes sur l’organisation 3D des chromosomes en mitose
Porteurs du project :
Pascal Bernard et Laurent Modolo (co-superviseurs)
Personnes impliquées :
Biologie expérimentale : Léonard Colin, Esther Toselli, Pascal Bernard
Bio-info : Arnaud Duvermy (étudiant candidate à la thèse), Laurent Modolo, Pascal Bernard.
Arnaud Duvermy est étudiant en bio-info à l’UBCL. Arnaud a déjà travaillé au sein de l’équipe Bernard lors d’un stage précédent, sous la co-supervision de Laurent et Pascal. Au cours de ce stage, Arnaud a notamment initié la création d’un pipeline d’analyse de ChIP-seq calibré.
Problématique et objectifs
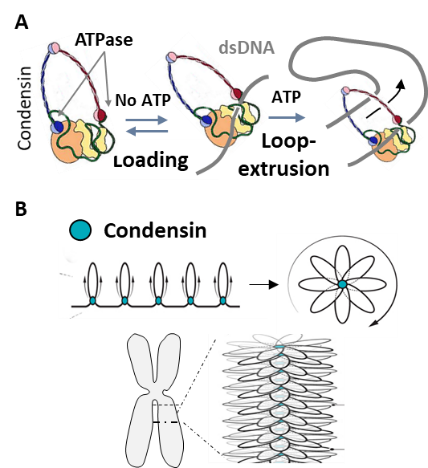 Condensine est un complexe organisateur du génome, pourvu d’une activité ADN-translocase ATP-dépendante, et connu principalement comme le motor moléculaire de l’assemblage des chromosomes mitotiques (Hassler et al., 2018; Hirano, 2016). Les mécanismes par lesquels condensine façonne les chromosomes mitotiques demeure mal compris. In vivo, des expériences d’Hi-C ont montré que condensine promeut la formation de boucles de chromatine de 80-100 kbp ainsi que des contacts à plus longue-distance (Gibcus et al., 2018; Kakui et al., 2017). In vitro, condensine se lie à une molécule d’ADN nu de façon électrostatique (indépendamment de la séquence AND), puis utilise l’énergie issue de cycles d’hydrolyse de l’ATP pour en former une boucle (Eeftens et al., 2016; Ganji et al., 2018; Kong et al., 2020). Les modèles actuels proposent que condensine organise le génome en 3D par un processus dit d’extrusion de boucles (Fig. 1A). La puissance de ce modèle tient en en sa capacité à expliquer l’organisation 3D des chromosomes par un unique mécanisme (Fig. 1B). Cependant deux aspects fondamentaux demeurent obscurs : (1) l’on ignore les détails structuraux et enzymologiques de la manipulation de l’ADN par condensine, et (2) l’on ignore également comment les réactions de liaison de condensine à l’ADN puis de formation d’une boucle s’opèrent dans le contexte encombré de génomes chromatinisés. L’objectif du project de thèse s’inscrit dans cette seconde problématique et vice à déterminer l’impact des nucléosomes sur le fonctionnement de condensine en utilisant la levure S. pombe comme organisme modèle.
Condensine est un complexe organisateur du génome, pourvu d’une activité ADN-translocase ATP-dépendante, et connu principalement comme le motor moléculaire de l’assemblage des chromosomes mitotiques (Hassler et al., 2018; Hirano, 2016). Les mécanismes par lesquels condensine façonne les chromosomes mitotiques demeure mal compris. In vivo, des expériences d’Hi-C ont montré que condensine promeut la formation de boucles de chromatine de 80-100 kbp ainsi que des contacts à plus longue-distance (Gibcus et al., 2018; Kakui et al., 2017). In vitro, condensine se lie à une molécule d’ADN nu de façon électrostatique (indépendamment de la séquence AND), puis utilise l’énergie issue de cycles d’hydrolyse de l’ATP pour en former une boucle (Eeftens et al., 2016; Ganji et al., 2018; Kong et al., 2020). Les modèles actuels proposent que condensine organise le génome en 3D par un processus dit d’extrusion de boucles (Fig. 1A). La puissance de ce modèle tient en en sa capacité à expliquer l’organisation 3D des chromosomes par un unique mécanisme (Fig. 1B). Cependant deux aspects fondamentaux demeurent obscurs : (1) l’on ignore les détails structuraux et enzymologiques de la manipulation de l’ADN par condensine, et (2) l’on ignore également comment les réactions de liaison de condensine à l’ADN puis de formation d’une boucle s’opèrent dans le contexte encombré de génomes chromatinisés. L’objectif du project de thèse s’inscrit dans cette seconde problématique et vice à déterminer l’impact des nucléosomes sur le fonctionnement de condensine en utilisant la levure S. pombe comme organisme modèle.
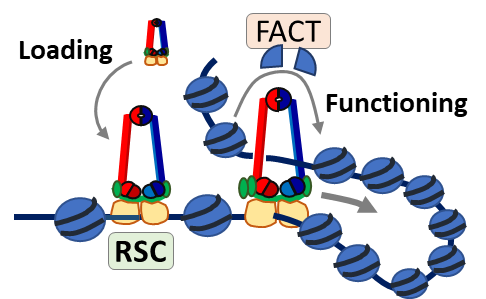 En combinant génétique classique des levures, imagerie chromosomique et génomique (MNase-seq) nous avons montré que condensine est enrichie à proximité de zones déplétées en nucléosomes en métaphase (Toselli-Mollereau et al., 2016). Nous avons également montré que l’association de condensine aux chromosomes est corrélée positivement à l’éviction des nucléosomes des promoteurs de gènes ; éviction opérée par Gcn5 et le complexe de remodelage RSC (Toselli-Mollereau et al., 2016). Nous avons proposé que les nucléosomes sont un obstacle pour la liaison de condensine à l’ADN, et que les promoteurs de gènes, déplétés en nucléosomes par l’action de RSC, constituent des sites privilégiés où condensine se lie à l’ADN génomique nu (Fig. 2 ; loading) (Robellet et al., 2017).
En combinant génétique classique des levures, imagerie chromosomique et génomique (MNase-seq) nous avons montré que condensine est enrichie à proximité de zones déplétées en nucléosomes en métaphase (Toselli-Mollereau et al., 2016). Nous avons également montré que l’association de condensine aux chromosomes est corrélée positivement à l’éviction des nucléosomes des promoteurs de gènes ; éviction opérée par Gcn5 et le complexe de remodelage RSC (Toselli-Mollereau et al., 2016). Nous avons proposé que les nucléosomes sont un obstacle pour la liaison de condensine à l’ADN, et que les promoteurs de gènes, déplétés en nucléosomes par l’action de RSC, constituent des sites privilégiés où condensine se lie à l’ADN génomique nu (Fig. 2 ; loading) (Robellet et al., 2017).
Qu’en est-il de l’impact des nucléosomes sur l’activité d’extrusion de boucles de condensine ?
En combinant des cribles génétiques et une approach non-biaisée de protéomique, nous avons identifié FACT et l’enzyme de remodelage Chd1 comme deux co-facteurs de condensine. FACT est une chaperonne d’histones bien connue pour faciliter la progression des ADN- et des ARN-polymérases le long de la fibre de chromatine en assurant le désassemblage/réassemblage des nucléosomes. ChD1 positionne les nucléosomes et collabore avec FACT à la fluidité de la chromatine. Nous spéculons que FACT et ChD1 sous-tendent l’activité de condensine en fluidifiant la chromatine durant la mitose (cf Fig.2 ; functioning).
Nous avons collecté des données préliminaires convergentes indiquant/suggérant : (1) que les nucléosomes inhibent le fonctionnement de condensine in vivo, (2) que condensine ne possède pas d’activité intrinsèque de remodelage de la chromatine lui permettant de franchir seule cette barrière nucléosomale, (3) que FACT prend part à la formation de boucles de chromatine en mitose, et (4) que l’activité de FACT affecte celle de condensine en mitose.
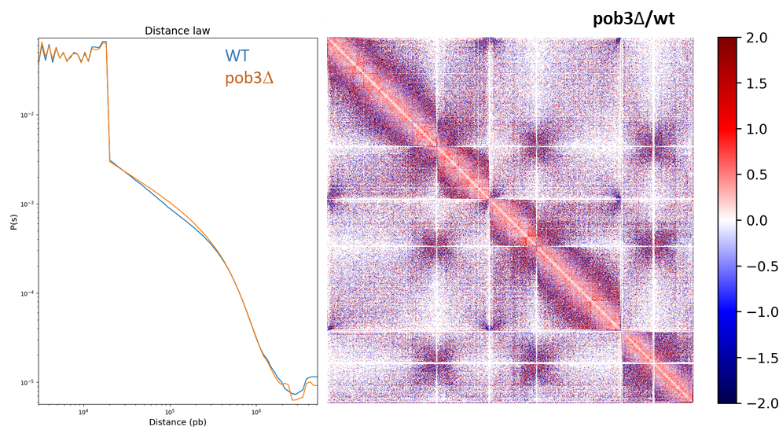 Léonard Colin a mis en place la technique de Hi-C dans l’équipe, et reproduit les données publiées de contacts 3D en métaphase (Kakui et al., 2017). De plus, nous observons dans les cellules mutantes FACT bloquées en métaphase une augmentation de la fréquence des contacts 3D à environ 100 kb de distance, i.e. dans la gamme des contacts mitotiques. Ceci, plus nos données génétiques liant FACT à condensine, suggère fortement que FACT prend part à la formation de boucles de chromatine par condensine, en mitose.
Léonard Colin a mis en place la technique de Hi-C dans l’équipe, et reproduit les données publiées de contacts 3D en métaphase (Kakui et al., 2017). De plus, nous observons dans les cellules mutantes FACT bloquées en métaphase une augmentation de la fréquence des contacts 3D à environ 100 kb de distance, i.e. dans la gamme des contacts mitotiques. Ceci, plus nos données génétiques liant FACT à condensine, suggère fortement que FACT prend part à la formation de boucles de chromatine par condensine, en mitose.
Le sujet de thèse proposé vice à déterminer comment le remodelage des nucléosomes par FACT impacte la formation de boucles de chromatine par condensine en mitose en déployant/utilisant, mais aussi en développant, des méthodes complémentaires d’analyse bio-informatiques.
Il s’agira notamment de :
(1) analyser les données de FACT, Condencine et RNA Pol issue de Chip-Seq calibré à l’aide de pipeline dédié mis en place au LBMC pendant son stage précédent.
(2) Développer un pipeline d’analyse dédié ainsi que développer une méthode de normalisation adaptée en absence de données de type INPUT pour analyser les données de Mnase-Seq calibrée. Nous avons développé une méthode de MNase-seq quantitative (calibrée) chez S. pombe et possédons les données de séquençage.
(3) Développer une pipeline d’analyse dédié pour analyser les données de Hi-C et confirmer le rôle de FACT dans la formation de boucles de chromatine en mitose. Il s’agira de créer un pipeline d’analyse Hi-C et d’analyser les données générées dans l’équipe.
(4) Développer une measure de la condensation des chromosomes par analyse d’image automatique de chromosomes dans des noyau de cellules uniques. Nous enregistrerons un empilement d’images de signaux ponctuels et fluorescents (l’un rogue et l’autre vert et distant de 5 Mb sur le chromosome II) dans le volume nucléaire en 3D.
(5) Mettre au point une analyse intégrative de type multi-omique pour analyser l’ensemble des données générées (condensine ChIP-seq ; nucléosomes MNase-seq, et Hi-C) afin de déterminer l’impact de FACT sur l’association de condensine à la chromatine ainsi que sur la position des nucléosomes en métaphase. Le but est d’utiliser une méthode de quantification des interactions spatiales entre les occurrences de ces différents signaux.
(1) Déterminer l’impact de FACT sur l’association de condensine à la chromatine. Pour cela l’étudiant devra utiliser, voire optimiser, le pipeline d’analyse de ChIP-seq quantitatif/calibré mis en place au LBMC pendant son stage précédent.
(2) Déterminer l’impact de FACT sur la position des nucléosomes en métaphase. Nous avons développé une méthode de MNase-seq quantitative (calibrée) chez S. pombe et possédons les données de séquençage. L’étudiant aura pour tâche de créer un pipeline d’analyse quantitative de MNase-seq et d’analyser les données. Notez que ce pipeline diffèrera du pipeline de ChIP-seq calibré notamment par l’absence des données de type INPUT.
3) De confirmer le rôle de FACT dans la formation de boucles de chromatine en mitose. Il s’agira de créer un pipeline d’analyse Hi-C et d’analyser les données générées dans l’équipe.
Notez que l’approche Hi-C (globale) sera adossée à une measure de la condensation des chromosomes par microscopie, sur cellules uniques. Cette seconde approach pourrait bénéficier de la création d’un programme d’analyse d’images dédié. Laurent : il s’agit de mesurer la condensation des chromosomes directement dans le noyau de cellules uniques en mesurant à partir d’un empilement d’image la distance physique séparant deux signaux chromosomiques, ponctuels et fluorescents (l’un rogue et l’autre vert et distant de 5 Mb sur le chromosome II) dans le volume nucléaire en 3D.
4) Croiser les données générées (condensine ChIP-seq ; nucléosomes MNase-seq, et Hi-C) afin de rechercher/identifier des corrélations spatiales. Pour cela l’étudiant pourra utiliser le logiciel HAWK Laurent ?
Données disponibles
Données MNase-seq calibrée (mutants condensine et FACT) : acquises et disponibles à l’analyse
Données de ChIP-seq calibré (Condensine et RNA Pol II): partiellement acquises, en cours de finalisation
Données de Hi-C en métaphase (mutants condensine et mutants FACT ou ChD1): partiellement acquises, en cours de finalisation
Début du project : Novembre 2021
Durée : 3 and