Avancement
- Début janvier 2023: récupération des données
Porteur du projet
Marie DELATTRE
Personnes
Ghislain DURIF et Laurent MODOLO
Problématique biologique
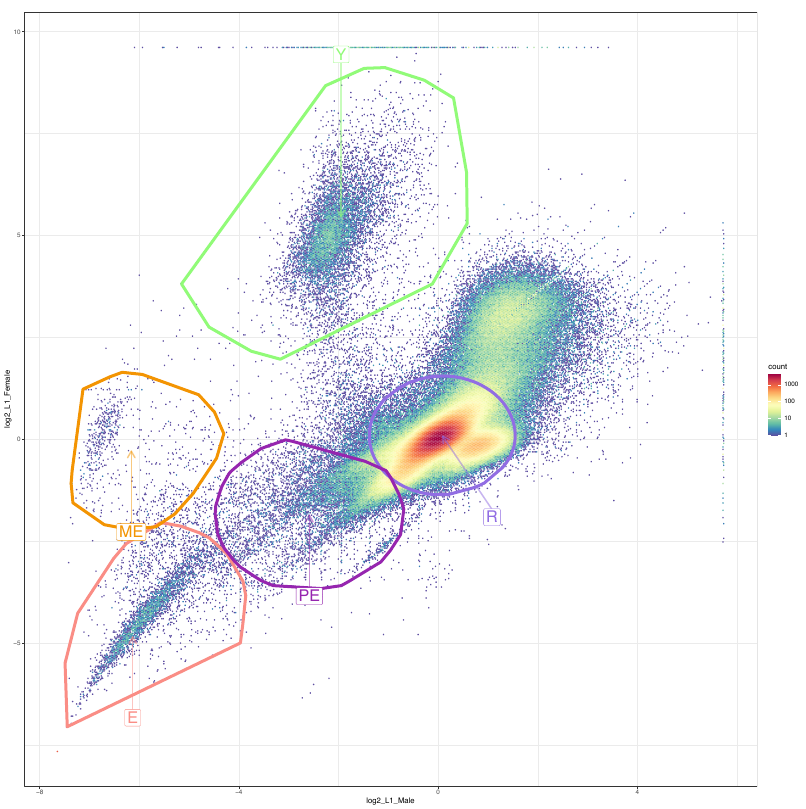
Nous étudions les nématodes du genre Mesorhabditis, qui subissent l’élimination programmée du génome (https://doi.org/10.1101/2022.03.19.484980). Toutes les cellules somatiques ont un génome réarrangé et réduit alors que le génome des cellules germinales est intact. Nous cherchons à simplifier l’identification des régions éliminées du génome dans le soma. Par la comparaison de couverture des reads DNAseq entre larves (possédant presque exclusivement du soma) et des adultes (possédant une lignée germinale développée, surtout chez les males), nous avons montré qu’il est possible d’identifier des clusters de séquence présents seulement chez les adultes (donc éliminés dans le soma _cluster E) ; présents uniquement chez les males (nous permettant de retrouver les séquences du chromosome Y_cluster Y) ; voire les séquences du Y étant éliminés dans le soma (absent chez les larves ; présent seulement chez les males adultes_cluster ME). Le cluster R correspond à l’ADN count retenu, présent aussi bien chez les adultes
Nous avons validé expérimentalement l’elimination de certaines de ces séquences, renforçant l’idée que ce clustering nous permet de bien identifier l’ADN éliminé.
Questions
Nous aimerions maintenant utiliser une approche statistique du problème pour mieux délimiter et valider les clusters. Également, nous pourrons tester quels sont les ratios exacts entre cellules somatiques et germinales dans les différents échantillons par l’analyse plus précise de ces clusters. Par exemple, nous savons qu’il y a une cellule germinale dans les larves et avons estimé 500 cellules somatiques. Notre estimation est vague et nous trouverons peut etre qu’elles en comportent en réalité 800. De la meme manière, la proportion germline/soma dans les adultes est basée sur des estimations grossières. Le positionnement attendu des clusters sur le graph en fonction des ratios pourra être testé.
Données
Pour l’espèce M. belari nous avons déjà toutes les données d’Illumina à très haute couverture (> 400X) pour des males et des femelles et des larves (voir Figure ci-dessus). L’analyse précédent a été réalisée en utilisant des fragments de 100 bp le long du génome et en calculant leur couverture dans chaque échantillon.
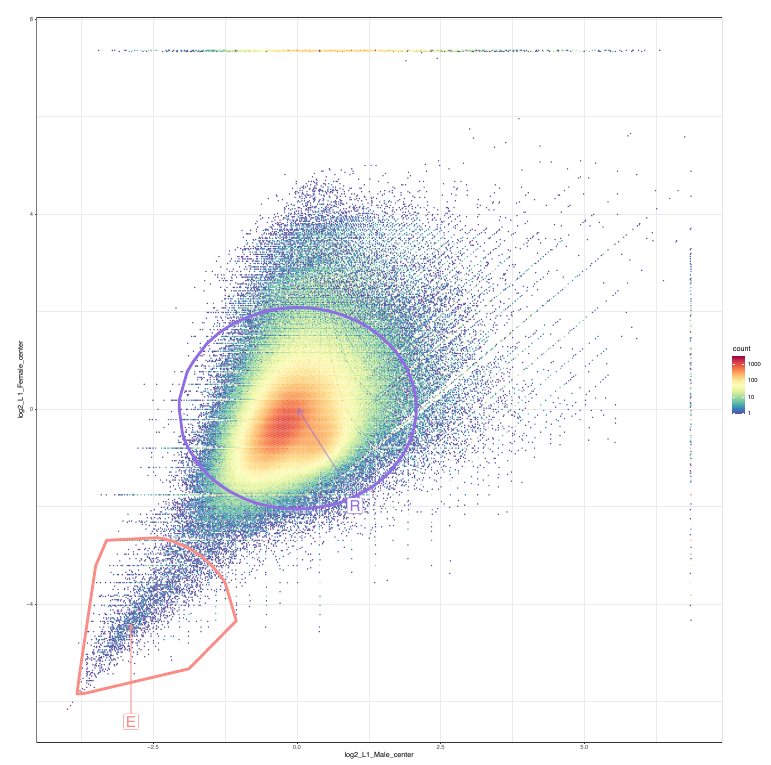
Nous avons également les mêmes données Illumina DNAseq pour l’espèce M. spiculigera dont un draft de génome existe également. Selon l’avancée du projet, ces données pourront aussi etre exploitées. Les données préliminaires (cf figure ci-contre) montre un clustering différent, en cohérence avec i) l’absence de chromosome Y dans cette espèce, 2) une proportion plus réduite d’ADN éliminé par rapport à M. belari (cluster E a priori plus réduit en taille que chez M. belari). S’il est nécessaire de valider l’analyse sur une troisième espèce, il y aura la possibilité de le faire sur M. monhystera pour laquelle le séquençage des larves, males et femelles est en cours.
Date
- date du début du projet: le plus tot possible
- date d’obtention des données: les données sont déjà disponibles
- date d’obtention de l’intégralité des données: -
- date souhaitée de fin du projet: premier semestre 2023 idéalement
Attentes
L’analyse initiale en cluster a été réalisée par Carine Rey, mais il n’y a pas eu d’analyse statistique associée. Valider cette approche nous sera très utile pour la suite du projet qui cherchera à identifier les séquences cibles de l’élimination dans un plus grand nombre d’espèces, tout en nous aidant à estimer les proportions de tissus (soma/germline) dans les différentes espèces.